열여덟번째 날: Perl과 생명정보학
저자
@ascendox - 현재 동국대학교 식물생명공학과 박사 과정으로 분자유전학실험실 유전육종학을 전공하고 있다. 프로필
시작하며
Perl은 문자열 처리 기능이 강력하기 때문에 문자열 구문 분석을 쉽게 할 수 있어 생명정보학에 종사하는 많은 사람들에게 인기가 높은 언어입니다. 특히 Perl이 가진 강력한 정규표현식 엔진 덕에 문자로 이루어진 서열(sequence; DNA:deoxyribonucleic acid, RNA: Ribonucleic acid, Protein) 분석에 중요한 역할을 하고 있습니다. 생물전공자가 이용할 만한 간단한 스크립트를 보면서 Perl이 생명정보학에서 어떻게 중요한 위치를 차지할 수 있게 되었는지를 생각해보는 문서가 되었으면 합니다.
생물체와 DNA 염기서열
보통 생물체의 유전정보는 DNA의 염기서열에 의해 결정된다고 합니다. DNA가 모여 하나의 기능을 하는 것을 유전자(gene)라고 하며, 유전자와 단백질(protein)이 합쳐진 것을 염색체(chromosome)라고 부르며 전체 염색체를 유전체(genome: gene + chromosome)라고 부릅니다. 이 유전체가 우리가 흔히 게놈 프로젝트라고 부를 때 앞에 있는 그 게놈입니다. :-)

그림 1. 벼에서 RING finger protein의 위치 (Perl 기반의 circos 사용; PMB 2010 72:369-380), (큰 그림을 보려면 클릭하세요)
생물학자(개인적으로 이 단어를 무척이나 싫어합니다만...)들이 자신이 발견한 혹은 수집한 유전자의 기능을 분석하기 위해 보통 가장 많이 사용하는 방법은 다른 유전자와 비교하는 것입니다. 이때 사용하는 방법 중 가장 대표적인 것이 BLAST(Basic Local Alignment Search Tool)입니다. BLAST의 NCBI 데이터베이스에 있는 DNA, 단백질 등의 정보를 이용해 서열(sequence)의 유사성(similarity)을 검색할 수 있습니다.
이런 류의 프로그램을 사용하기 위해서는 입력 데이터가 필요합니다. 물론 DNA,RNA, 단백질에 대한 정보일텐데 특정한 형식이어야 할 것입니다. 대부분의 서열 검색 및 분석 프로그램, 데이터베이스는 서열을 받아 들이기 위한 입력 형식으로 FASTA 형식을 사용합니다.
FASTA 형식이란?
생명정보학에서 FASTA 형식은 텍스트 기반의 형식으로 뉴클레오티드(핵산의 구성 성분) 서열이나 펩티드(두 개 이상의 아미노산 분자로 이뤄지는 화학 물질) 서열을 표현하기 위한 형식입니다. 문자열 기반인 FASTA 형식은 DNA, RNA, 단백질 정보를 포함하며 염기쌍(base pair)이나 아미노산을 한 글자 코드를 사용해서 표현합니다. 문자열 기반인 FASTA 형식은 단순하기 때문에 문자열 처리 도구나, Perl과 같은 스크립트 언어를 사용해서 서열 정보를 생성하거나 파싱하기 쉽습니다.
다음은 DNA 서열의 예입니다.
1 2 3 4 5 6 | >DNA_SEQUENCE_1ATGGGAGATAGCGACTACGACTACGACTACGACTACGACTACGACTGACGACTGATCATCTACTCTACTACTGATGCACTGACTGATTACTACTACTATCTGACGACTACTATACGACAGCATGCTATATAATATAAGCACACACTACGACTACTATGCTGCTACGACTGACTGATGTGACTGACATCATCTAATATATATATATATAGCATCAGCATCACGATCACGACTACGACTACGACTAAATCACTACGACTCGCATCACTAG |
다음은 단백질 서열의 예입니다.
1 2 3 | >Protein_SEQUENCE_2SATVSEINSETDFVAKNDQFIALTKDTTAHIQSNSLQSVEELHSSTINGVKFEEYLKSQIATIGENLVVRRFATLKAGANGVVNGYIHTNGRVGVVIAAACDSAEVASKSRDLLRQICMH |
FASTA 형식은 서열 앞 부분에 서열 이름이나 주석을 넣을 수도 있습니다.
즉 앞의 예제에서 볼 수 있듯이 > 다음에 유전자 이름 정보(header)를
적고 다음 줄부터 서열 정보를 기입하는 정말 간단한 구조를 가지고 있습니다.
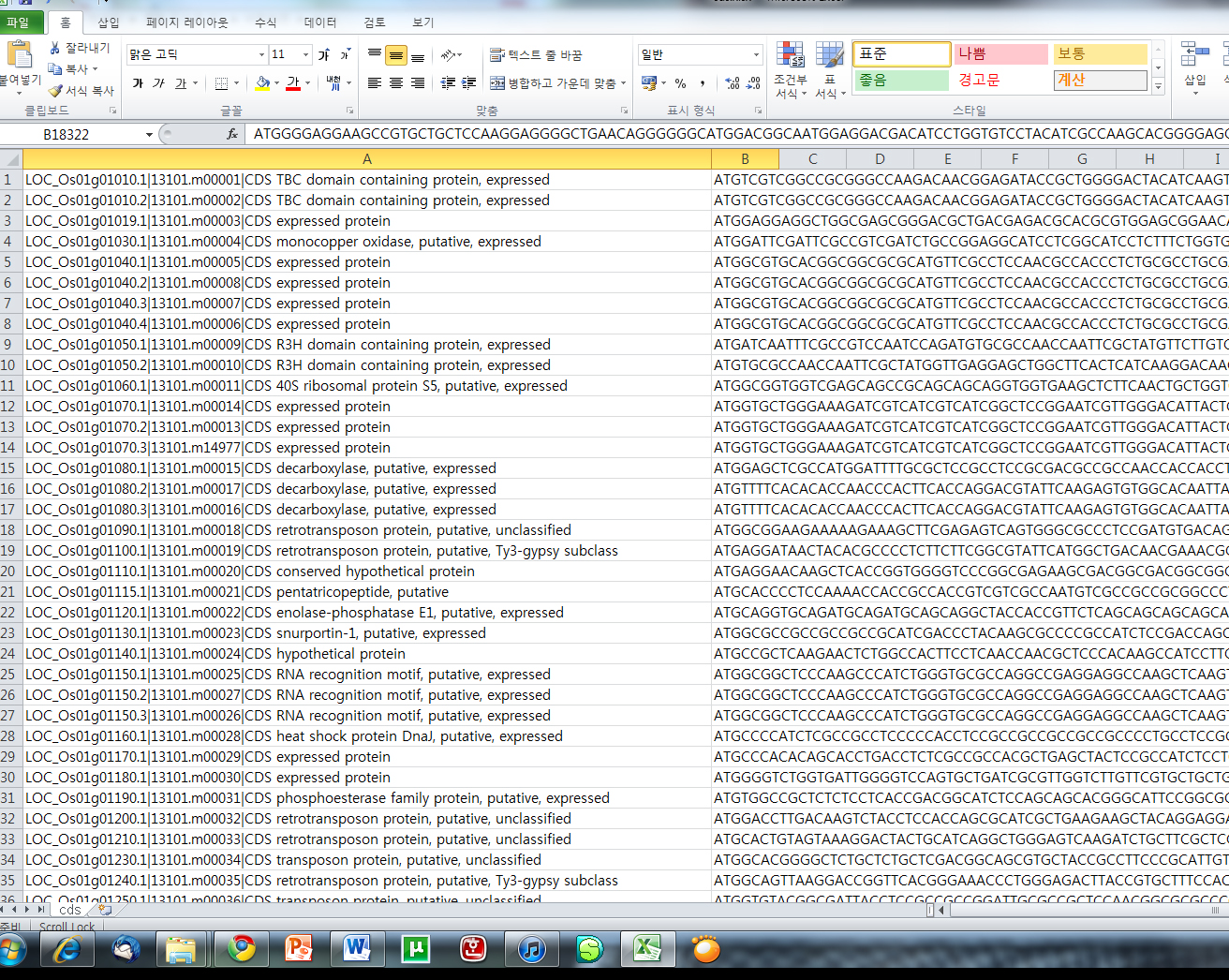
하지만 서열 정보를 보관할 때 파스타 형식으로 보관하면 수만 개의 서열 정보를 찾기에도 어려울 뿐더러 뽑아내기도 쉽지 않습니다. 자료의 2차 가공을 위해 보통은 엑셀로 옮긴 다음 파일을 정리합니다. 다음은 FASTA 형식을 엑셀로 옮긴 화면입니다. A열에는 유전자 이름이 B열에는 서열을 저장했습니다.
반대로 서열 정보를 가지고 있는 사이트는 대부분 FASTA 형식으로 서열 정보를 제공하는 경우가 대부분이라 엑셀로 변환하기가 쉽지않으며, 또한 사이트를 사용할 때는 다시 FASTA 형식으로 변환해야하기 때문에 수천개의 서열을 바꾸는 것을 손으로 한다면 하루가 꼬박 걸리는 노가다성 작업입니다.
지금 필요한 것은 무엇? Perl!
FASTA 형식을 엑셀에서 읽을 수 있도록 탭을 구분자로 하는 CSV 형식의 파일로
또는 반대의 형식으로 변환할 때 Perl을 이용하면 매우 편리할 것입니다.
FASTA 형식을 탭 구분자 형식의 파일로 변경하는 fasta2tab.pl 스크립트는
다음과 같습니다.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 | #!usr/bin/env perl## Usage:# perl fasta2tab.pl <src_file>#use 5.010;use strict;use warnings;use autodie;# Parses FASTA files into tab-delimited files.# The result is of the format:# SEQNAME tab SEQDESCRIPTION tab SEQUENCEmy @header;my @sequence;my @result;LINE:while (<>) { state $reading_seq; chomp; if ( $reading_seq ) { if ( /^>/ ) { # already reading a sequence but there is a header # on this line, so stop reading, make an entry in # the result list, clear the sequence list and redo # that line push @result, { header => \@header, sequence => \@sequence, }; undef @sequence; undef $reading_seq; redo LINE; } else { # already reading a sequence and no new header on # this line, so remove whitespace and add the line # to the currently read sequence s/\s+//g; push @sequence, $_; } } else { if ( /^>/ ) { # found the start of a sequence, # so get the header and shift off the > character s/^>\s*//; @header = split; $reading_seq = 1; next LINE; } }}# add the last sequence to the result list when# we have run out of lines in the source file as# this is the only one whose end is not delimited# by the start of a new sequenceif ( @header && @sequence ) { push @result, { header => \@header, sequence => \@sequence, };}# print the result to the stdoutfor ( @result ) { my $seq_name = shift @{ $_->{header} }; my $seq_desc = join q{ }, @{ $_->{header} }; my $sequence = join q{}, @{ $_->{sequence} }; say join "\t", $seq_name, $seq_desc, $sequence;} |
탭 구분자 형식의 파일에서 FASTA 형식의 파일로 변경하는
tab2fasta.pl 스크립트는 다음과 같습니다.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | #!/usr/bin/env perl## Usage:# perl tab2fasta.pl <src_file1> [<src_file2> ... ]#use 5.010;use strict;use warnings;use autodie;use Text::Wrap;$Text::Wrap::columns = 72;while (<>) { chomp; my ( $seq_name, $seq_desc, $sequence ) = split "\t"; say ">$seq_name $seq_desc"; say wrap($sequence);} |
정리하며
Perl에는 생물학자를 위한 강력한 라이브러리인 BioPerl이 있습니다(물론 BioPython, BioJava 등도 있습니다). Bioperl을 이용하면 여러 종류의 입력 형식을 이용해서 서열을 변경하고 출력물을 정리 할 수 있습니다. BLAST, PAML, ClustalX 등 어지간한 프로그램 관련 모듈은 이미 안정적으로 잘 지원하고 있으며, 문자열 구분 분석 계산 스크립트도 응용되어 사용되고 있습니다. 워낙 방대한 기능을 제공하기도 하고, 문서 정리도 잘되어 있으므로 BioPerl 라이브러리를 살펴보는 것 역시 잊지 마세요.
실용적인 차원에서 Perl을 쓰고 있는지라 복잡하고 아름다운 코드를 보여드리지는 못하지만, 그럼에도 불구하고 최소한의 노력으로도 꽤 그럴듯한 결과를 내주는 것이 Perl이란 언어입니다. 덕분에 생물학 분야에서 Perl은 여전히 중요하게 사용되고 있으며, 앞으로 Personal Genome 시대인만큼 Perl에 대한 더 많은 수요가 생길 것이라 생각됩니다.
 거침없이 배우는 펄
거침없이 배우는 펄